על תסמונת רט

על תסמונת רט
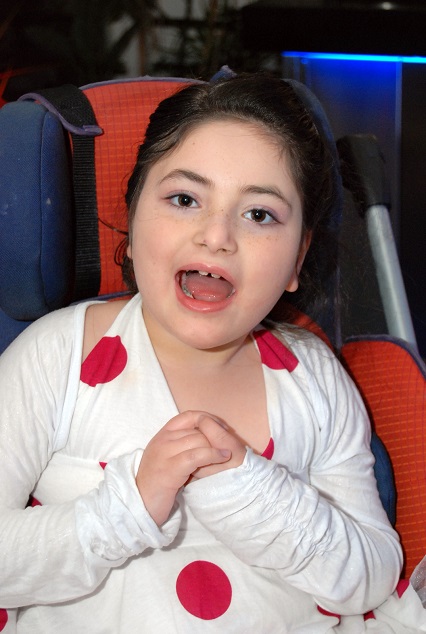
תסמונת רט היא הפרעה נוירו-גנטית התפתחותית שאינה תורשתית המופיעה בעיקר בבנות (1 לכ- 10,000 לידות של בנות). התסמונת מתאפיינת בהתפתחות לכאורה תקינה בשלבי החיים המוקדמים, שאחריה מופיע מגוון הפרעות: איבוד יכולת הדיבור, אובדן יכולות השימוש בידיים, הפרעות בהליכה, נשימה לא סדירה וקשיי עיכול. למרות הקשיים איתם הן מתמודדות, ילדות הלוקות בתסמונת רט בעלות עיניים חכמות ו"מדברות". הן יוצרות קשר עם סביבתן בעזרת עיניהן. הן פעילות, חיוניות ומלאות שמחת חיים ומסיבה זו מכונות בחיבה ברחבי העולם "מלאכיות הדממה".


| 1999 |
נמצא כי מוטציות בגן MECP2 שבכרומוזום ה-X הן הגורם לתסמונת רט |
חוקרים מפתחים מודל של עכברי רט ומתחיל המחקר בעכברים | 2001 |
| 2007 |
מתגלה פריצת דרך מחקרית |
מתחילים מחקרים קליניים לראשונה בעולם | 2012 |
| 2013 |
במחקר נוסף, שהעמותה שותפה במימונו, מצאה ד"ר מנדל מאורגון, ארה"ב שגנים תקינים |
בימים אלה מתנהלים מחקרים קליניים | 2016 |
| 2017 |
התחילה תוכנית תלת שנתית לריפוי התסמונת |
חברת הביוטק Avexis מתכננת להתחיל ניסוי קליני בריפוי גנטי בבני אדם | 2018 |